# Title : 16082019 Script for the visualization of the correlation between number of common interactions and type of interactions
import pandas as pd
import numpy as np
from collections import defaultdict
import seaborn as sns
import matplotlib.pyplot as plt
from statistics import mean
%matplotlib inline
def query_interactions(query):
d2 = defaultdict(dict)
data=pd.read_excel(r'C:\Users\linigodelacruz\Documents\PhD_2018\Documentation\Calculations\data_sgd\Interaction_data_sgd_downloads.xlsx',header=17,encoding="utf-8-sig")
col_label=data.columns.values
#query=['CLN1']
# giant for loop
names1 = query
i=-1
for query1 in names1:
#filtering the table just for the value of the query
q1 = data[data['Standard_Gene_Name_(Bait)']==query1]
#r=get_row(query1)
#df1 = pd.DataFrame(q1, columns=["excel_number", "Interactors of query1"])
#gb1 = df1.groupby("Interactors of query1", sort=False) # the grouping reorganizes the set alphatically, the sort by default is true!!!!!!
#gb1_row = df1.groupby("excel_number",sort=False)
q1_interact=q1[col_label[3]].unique()
# a for loop for all the interactors of query
for query2 in q1_interact:
#i+=1
# write in a matrix , where the 1st column are the names in q1 and the first row = q1[0]+get_query(q1[0])
q2=data[data['Standard_Gene_Name_(Bait)']==query2] #these are get_query(q1[i])
q2_interact=q2[col_label[3]].unique()
# how to avoid that df2 overlaps with the next value?
#df2 = pd.DataFrame(q2, columns=["excel raw number of q2", "Interactors of query2"])
#gb2 = df2.groupby("Interactors of query2",sort=False)
#names3 = [name3 for name3, row2 in gb2] # to visualize what is in the interactors for query 2
d = defaultdict(int)
common = []
for name1 in q2_interact:
if name1 in q1_interact: # if a gene interactor of the query1 is in interactors of query 2
common.append(name1)
d[name1] += 1
d2[query1, query2]["common"] = common
d2[query1,query2]["names of genes"]=query2
d2[query1, query2]["n_common"] = len(common)
d2[query1, query2]["query gene info length"] = len(q1_interact)
d2[query1, query2]["interactors of query gene info length"] = len(q2_interact)
if len(q1)==0:
d2[query1, query2]["% of query subset"] = 0
else:
d2[query1, query2]["% of query subset"] = len(d)/len(q1_interact) *100
if len(q2)==0:
d2[query1, query2]["% of query 2 subset "] = 0
else:
d2[query1, query2]["% of query 2 subset "] = len(d)/len(q2_interact) *100
q1_filt=q1[q1[col_label[3]]==query2]
# if q1_filt[col_label[4]].any()=='Synthetic Lethality ':
interactions_lethality=(q1_filt[col_label[4]]=='Synthetic Lethality')
interactions_negative=(q1_filt[col_label[4]]=='Negative Genetic')
interactions_positive=(q1_filt[col_label[4]]=='Positive Genetic')
d2[query1,query2]["Type"]= 'not clear interaction'
if interactions_lethality.any()==True:
d2[query1,query2]["Type"]= 'Synthetic Lethality'
#else:
# if interactions_lethality.all()==False and interactions_negative.any()==False and interactions_positive.any()==False:
#d2[query1,query2]["Type"]='Not clear'
if interactions_negative.all()==True:
d2[query1,query2]["Type"]= 'Negative'
#else:
# if interactions_lethality.all()==False and interactions_negative.any()==False and interactions_positive.any()==False:
#d2[query1,query2]["Type"]='Not clear'
if interactions_positive.all()==True:
d2[query1,query2]["Type"]= 'Positive'
#else:
# if interactions_lethality.all()==False and interactions_negative.any()==False and interactions_positive.any()==False:
#d2[query1,query2]["Type"]='Not clear'
# else:
d2[query1,query2]["interact_annotation"]=q1_filt[col_label[4]]
df=pd.DataFrame(d2).T
df_sorted=df.sort_values(by=['% of query subset'])
df_sorted=df_sorted[::-1]
df_sorted.to_excel("data_output_"+ "".join(query) + ".xlsx")
data=pd.read_excel(r'C:\Users\linigodelacruz\Documents\PhD_2018\Documentation\Calculations\Functions\data_output_'+ "".join(query) + ".xlsx")
return data
genes_passed=[['NRP1'],['BEM3'],['GPR1'],['ACT1'],['BRE5'],['CDC28'],['HSP82'],['CDC3'],['ADE13'],['SEC14'],['ARP2'],['BIM1'],
['BEM4'], ['BUB3'],['CNB1'],['GIM3'],['LTE1'],['MAD1'],['PHO85'],['SEC15'],['SEC3'],['SMI1'],['SMY1'],['SWI4'],
['BEM2'],['BEM1'], ['CDC24'],['CDC42'],['RDI1'],['CLA4'],['RSR1'],['RGA1'],['BNI1'],['GIC2'],['BNR1'],
['MYO1'],['MSB3'],['EXO70'],['CLN1'],['CLN2'],['IRA2'],['WHI2'],['WHI3']]
data_test=[]
for names in genes_passed:
output=query_interactions(names)
data_test.append(output)
## Functions
def positive_int(data):
return data[data['Type']=='Positive']['% of query subset']
def positive_query2(data):
return data[data['Type']=='Positive']['% of query 2 subset ']
def negative_int(data):
return data[data['Type']=='Negative']['% of query subset']
def negative_query2(data):
return data[data['Type']=='Negative']['% of query 2 subset ']
def lethal_int(data):
return data[data['Type']=='Synthetic Lethality']['% of query subset']
def lethal_query2(data):
return data[data['Type']=='Synthetic Lethality']['% of query 2 subset ']
data=data_test
## Positive Interactions
data_positive=[]
data_positive_query2=[]
for names in data:
data_positive.append(positive_int(names))
data_positive_query2.append(positive_query2(names))
## Negative Interactions
data_negative=[]
data_negative_query2=[]
for names in data:
data_negative.append(negative_int(names))
data_negative_query2.append(negative_query2(names))
## Synthetic Lethal interactions
data_lethality=[]
data_lethality_query2=[]
for names in data:
data_lethality.append(lethal_int(names))
data_lethality_query2.append(lethal_query2(names))
len(data)
43
## Average
mean_values_positive=[]
mean_values_positive_query2=[]
std_values_positive=[]
std_values_positive_query2=[]
mean_values_negative=[]
std_values_negative=[]
mean_values_negative_query2=[]
std_values_negative_query2=[]
mean_values_lethal=[]
std_values_lethal=[]
mean_values_lethal_query2=[]
std_values_lethal_query2=[]
for i in range(0,len(data)):
mean_values_positive.append(np.mean(data_positive[i]))
mean_values_positive_query2.append(np.mean(data_positive_query2[i]))
std_values_positive.append(np.std(data_positive[i]))
std_values_positive_query2.append(np.std(data_positive_query2[i]))
mean_values_negative.append(np.mean(data_negative[i]))
std_values_negative.append(np.std(data_negative[i]))
mean_values_negative_query2.append(np.mean(data_negative_query2[i]))
std_values_negative_query2.append(np.std(data_negative_query2[i]))
mean_values_lethal.append(np.mean(data_lethality[i]))
std_values_lethal.append(np.std(data_lethality[i]))
mean_values_lethal_query2.append(np.mean(data_lethality_query2[i]))
std_values_lethal_query2.append(np.std(data_lethality_query2[i]))
mean_values=[mean_values_positive,mean_values_negative,mean_values_lethal]
mean_values_query2=[mean_values_positive_query2,mean_values_negative_query2,mean_values_lethal_query2]
std_values=[std_values_positive,std_values_negative,std_values_lethal]
std_values_query2=[std_values_positive_query2,std_values_negative_query2,std_values_lethal_query2]
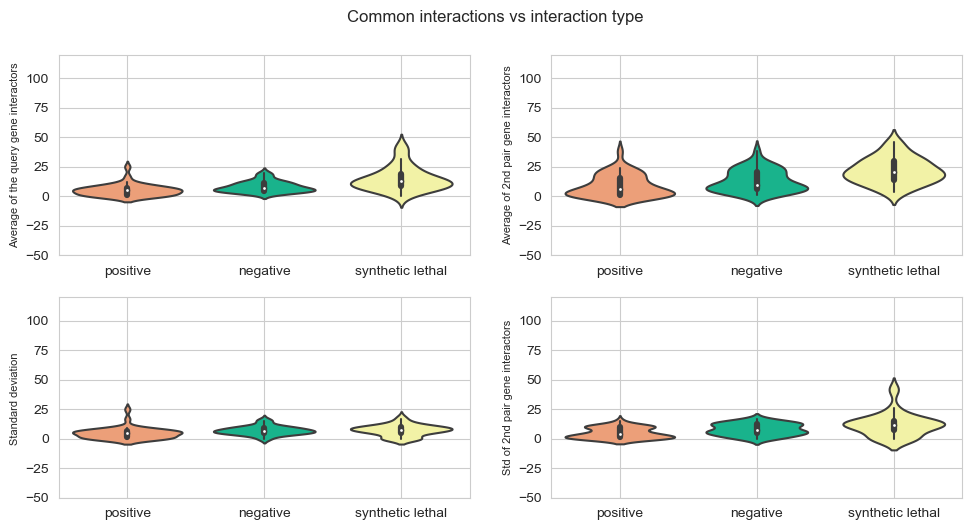
## Violin plot for the averages and std for each gene positive, negative and synthetic lethal interactions
## Data quality
# Make a dictionary with one specific color per group:
my_pal = {"#ff9966", '#ffff99', '#00cc99'}
fig, axes = plt.subplots(2, 2, figsize=(10,5), dpi=100, sharex=True, sharey=True)
plt.subplots_adjust(bottom=0.2, right=0.5, top=1.4)
fig.suptitle('Common interactions vs interaction type',x=0.5,y=1.05)
plt.subplot(2,2, 1)
sns.violinplot(data=mean_values,scale="count",palette = my_pal)
plt.xticks(np.arange(0,3),['positive','negative','synthetic lethal'])
plt.ylabel('Average of the query gene interactors',{'fontname':'Arial', 'size':'8'})
plt.grid(True)
plt.ylim(-50,120)
plt.subplot(2,2,2)
#sns.stripplot(data=mean_values,palette = my_pal)
sns.violinplot(data=mean_values_query2,scale="count",palette = my_pal)
plt.xticks(np.arange(0,3),['positive','negative','synthetic lethal'])
plt.grid(True)
plt.ylim(-50,120)
plt.ylabel('Average of 2nd pair gene interactors',{'fontname':'Arial', 'size':'8'})
plt.subplot(2,2, 3)
sns.violinplot(data=std_values,scale="count",palette = my_pal)
plt.xticks(np.arange(0,3),['positive','negative','synthetic lethal'])
plt.ylabel('Standard deviation ',{'fontname':'Arial', 'size':'8'})
plt.grid(True)
plt.ylim(-50,120)
plt.subplot(2,2,4)
#sns.stripplot(data=std_values,palette = my_pal)
sns.violinplot(data=std_values_query2,scale="count",palette = my_pal)
plt.xticks(np.arange(0,3),['positive','negative','synthetic lethal'])
plt.grid(True)
plt.ylim(-50,120)
plt.ylabel('Std of 2nd pair gene interactors',{'fontname':'Arial', 'size':'8'})
plt.tight_layout()
plt.savefig("violinplot-average-std-common-interactors-vs-interaction-type.svg",dpi=300,format='svg')
plt.savefig("violinplot-average-std-common-interactors-vs-interaction-type.png",dpi=300,format='png')
C:\Users\linigodelacruz\AppData\Local\Continuum\anaconda3\lib\site-packages\scipy\stats\stats.py:1713: FutureWarning: Using a non-tuple sequence for multidimensional indexing is deprecated; use `arr[tuple(seq)]` instead of `arr[seq]`. In the future this will be interpreted as an array index, `arr[np.array(seq)]`, which will result either in an error or a different result.
return np.add.reduce(sorted[indexer] * weights, axis=axis) / sumval
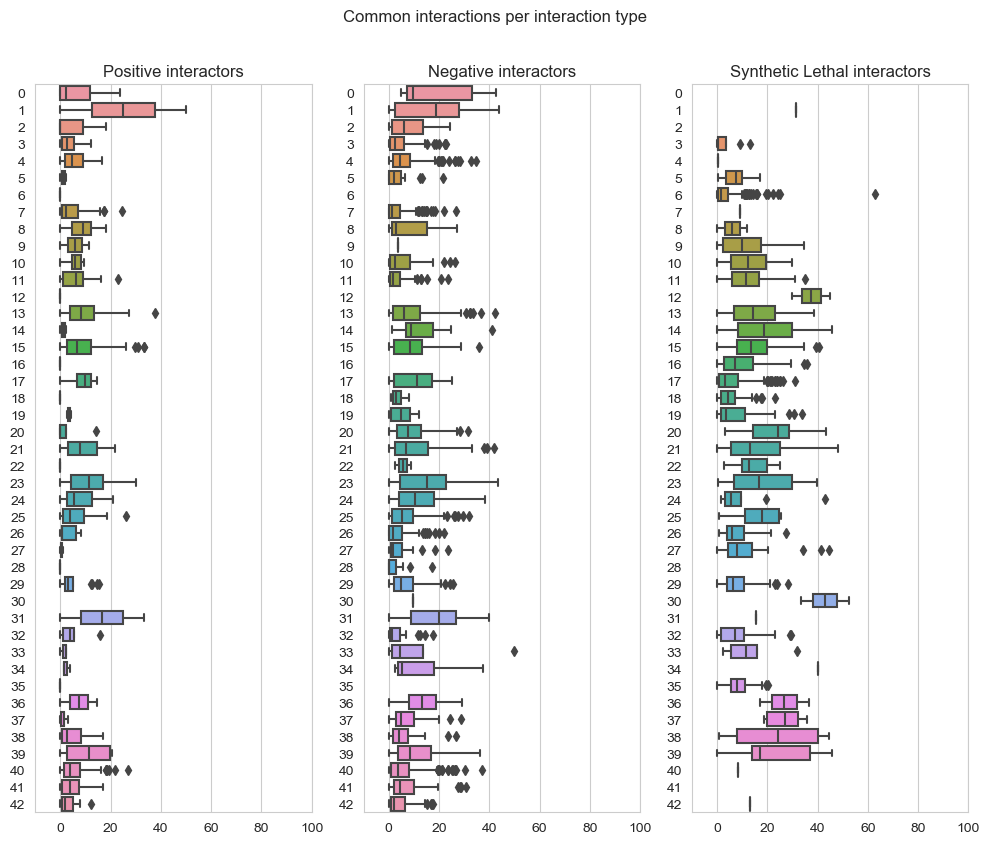
fig, axes = plt.subplots(1, 3, figsize=(10,8), dpi=100, sharex=True, sharey=True)
fig.suptitle('Common interactions per interaction type',x=0.5,y=1.05)
plt.subplot(1,3, 1)
plt.title('Positive interactors')
plt.grid(True)
plt.ylabel('')
plt.xlim(-10,100)
ax = sns.boxplot(data=data_positive,orient="h")
sns.set_style("whitegrid")
plt.subplot(1,3, 2)
plt.title('Negative interactors')
plt.grid(True)
plt.xlim(-10,100)
ax1 = sns.boxplot(data=data_negative,orient="h")
sns.set_style("whitegrid")
plt.subplot(1,3,3)
plt.title('Synthetic Lethal interactors')
plt.grid(True)
plt.xlim(-10,100)
ax2 = sns.boxplot(data=data_lethality,orient="h")
sns.set_style("whitegrid")
plt.subplots_adjust(bottom=0.5, right=1.4, top=1.4)
plt.tight_layout()
plt.savefig("boxplot-common-interactors-vs-interaction-type.tiff",dpi=300,format='tiff')
plt.savefig("boxplot-common-interactors-vs-interaction-type.png",dpi=300,format='png')
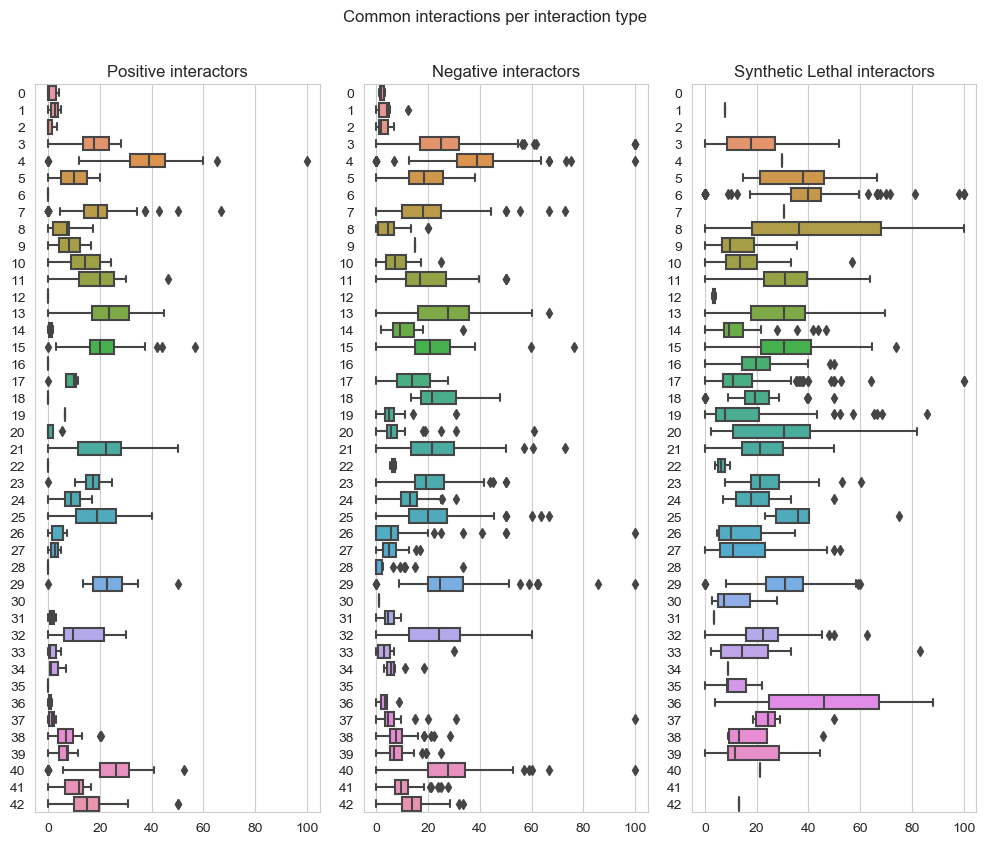
fig, axes = plt.subplots(1, 3, figsize=(10,8), dpi=100, sharex=True, sharey=True)
fig.suptitle('Common interactions per interaction type',x=0.5,y=1.05)
plt.subplot(1,3, 1)
plt.title('Positive interactors')
plt.grid(True)
plt.ylabel('')
plt.ylim(0,100)
ax = sns.boxplot(data=data_positive_query2,orient="h")
sns.set_style("whitegrid")
plt.subplot(1,3, 2)
plt.title('Negative interactors')
plt.grid(True)
plt.ylim(0,100)
ax1 = sns.boxplot(data=data_negative_query2,orient="h")
sns.set_style("whitegrid")
plt.subplot(1,3,3)
plt.title('Synthetic Lethal interactors')
plt.grid(True)
plt.ylim(0,100)
ax2 = sns.boxplot(data=data_lethality_query2,orient="h")
sns.set_style("whitegrid")
plt.subplots_adjust(bottom=0.5, right=1.4, top=1.4)
plt.tight_layout()
plt.savefig("boxplot-2nd-pair-common-interactors-vs-interaction-type.tiff",dpi=300,format='tiff')
plt.savefig("boxplot-2nd-pair-common-interactors-vs-interaction-type.png",dpi=300,format='png')
## Plots histogram
fig, axes = plt.subplots(1, 3, figsize=(10,5), dpi=100, sharex=True, sharey=True)
fig.suptitle('Common interactions vs interaction type',x=0.5,y=1.05)
kwargs_positive = dict(alpha=0.5, bins=20, density=True, stacked=True)
kwargs_negative = dict(alpha=0.5, bins=20, density=True, stacked=True)
kwargs_lethal = dict(alpha=0.5, bins=20, density=True, stacked=True)
# Set the font dictionaries (for plot title and axis titles)
title_font = {'fontname':'Arial', 'size':'16', 'color':'black', 'weight':'normal',
'verticalalignment':'bottom'} # Bottom vertical alignment for more space
axis_font = {'fontname':'Arial', 'size':'8'}
## Positive Interactions
plt.subplots_adjust(bottom=0.5, right=1.4, top=1.4)
#fig.suptitle('This is a somewhat long figure title', fontsize=16)
plt.subplot(1,3, 1)
plt.grid(True)
plt.ylabel('Normalized frequency',**axis_font)
plt.xlabel('% of common interactions with the query gene',**axis_font)
plt.xlim(0,100)
plt.ylim(0,0.2)
plt.hist(data_positive_query2[1:len(data_positive_query2)],**kwargs_positive,label=['Positive Interactions'],cumulative=False)
plt.title('Positive Interactions')
plt.subplot(1,3, 2)
plt.grid(True)
#plt.ylabel('Normalized frequency',**axis_font)
plt.xlim(0,100)
plt.ylim(0,0.2)
plt.xlabel('% of common interactions with the query gene',**axis_font)
plt.hist(data_negative_query2[1:len(data_negative_query2)],**kwargs_negative,label=['Negative Interactions'],cumulative=False)
plt.title('Negative Interactions')
plt.subplot(1,3, 3)
plt.grid(True)
#plt.ylabel('Normalized frequency',**axis_font)
plt.xlim(0,100)
plt.ylim(0,0.2)
plt.xlabel('% of common interactions with the query gene',**axis_font)
plt.hist(data_lethality_query2[1:len(data_lethality_query2)],**kwargs_lethal,label=['Lethality Interactions'],cumulative=False)
plt.title('Synthetic Lethal Interactions')
plt.tight_layout()
plt.savefig("common-interactors-vs-interaction-type.tiff",dpi=300,format='tiff')
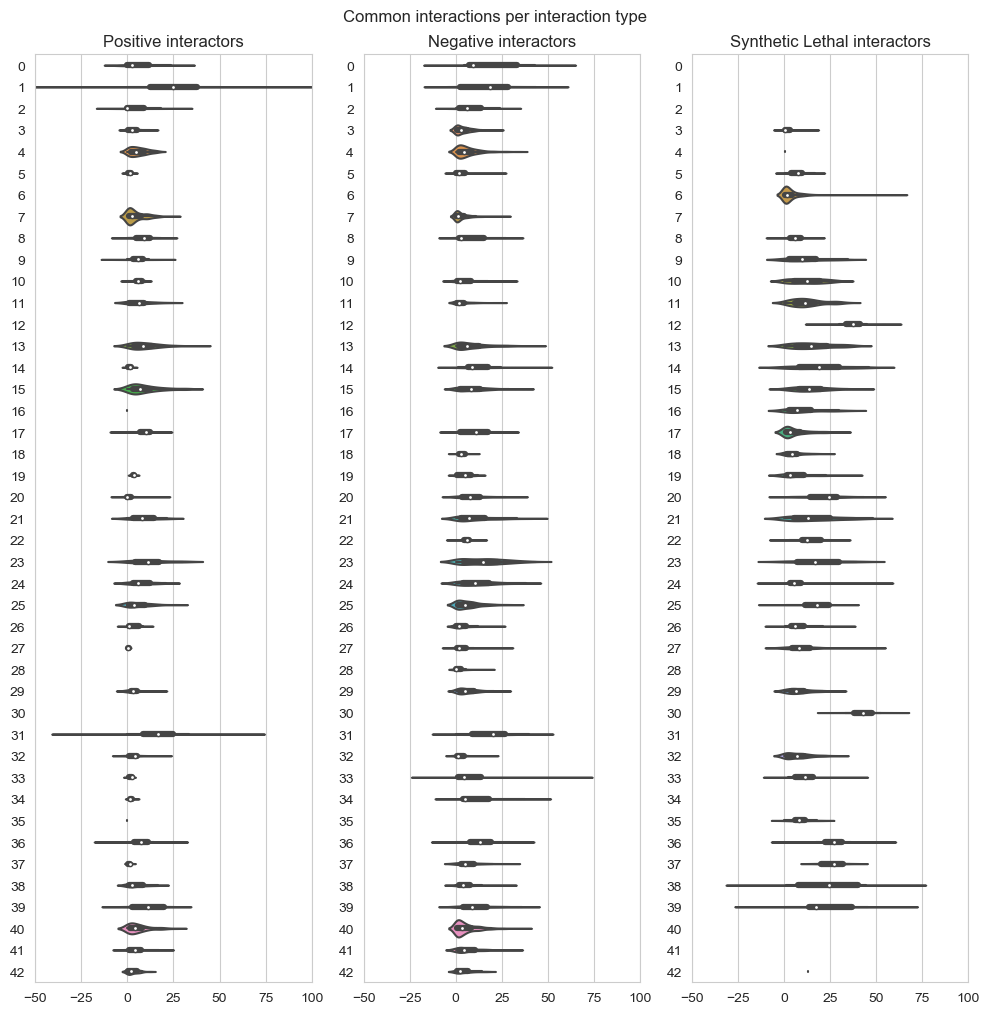
fig, axes = plt.subplots(1, 3, figsize=(10,10), dpi=100, sharex=True, sharey=True)
fig.suptitle('Common interactions per interaction type',x=0.5,y=1.01)
plt.subplot(1,3, 1)
plt.title('Positive interactors')
plt.grid(True)
plt.ylabel('')
plt.xlim(-50,100)
ax = sns.violinplot(data=data_positive,scale="count",orient='h')
sns.set_style("whitegrid")
plt.subplot(1,3, 2)
plt.title('Negative interactors')
plt.grid(True)
plt.xlim(-50,100)
ax1 = sns.violinplot(data=data_negative,scale="count",orient='h')
sns.set_style("whitegrid")
plt.subplot(1,3,3)
plt.title('Synthetic Lethal interactors')
plt.grid(True)
plt.xlim(-50,100)
ax2 = sns.violinplot(data=data_lethality,scale="count",orient='h')
sns.set_style("whitegrid")
plt.subplots_adjust(bottom=0.5, right=1.4, top=1.4)
plt.tight_layout()
plt.savefig("violinplot-common-interactors-vs-interaction-type.tiff",dpi=300,format='tiff')
C:\Users\linigodelacruz\AppData\Local\Continuum\anaconda3\lib\site-packages\scipy\stats\stats.py:1713: FutureWarning: Using a non-tuple sequence for multidimensional indexing is deprecated; use `arr[tuple(seq)]` instead of `arr[seq]`. In the future this will be interpreted as an array index, `arr[np.array(seq)]`, which will result either in an error or a different result.
return np.add.reduce(sorted[indexer] * weights, axis=axis) / sumval