
DNA extraction from SATAY samples
Contents
35. DNA extraction from SATAY samples#
35.1. Date#
19082020-24082020
35.2. Objective#
To extract the DNA from the cell pellets to further prepare for sequencing.
35.3. Method#
Genomic DNA extraction and enzymatic lisis (https://www.elabjournal.com/members/protocol/?protID=58618)1
Sorbitol solution:
0.9 M Sorbitol
0.1 M EDTA (pH 8.5)
Preparation:
Final volume : 50mL
0.9 x 182.17 g/mol /20 (0.9M)=8.2g Sorbitol
10mL 0.5M EDTA
Fill with MiliQ until 45mL and measure pH and adjust it to 8.5, in this case it was 7.2 so we needed to add roughly 6 drops of NaOH to reach 8.5.
Complete the 50ml with MiliQ.
10xTE:
100 mM Tris (pH 7.5)
10 mM EDTA
Preparation:
Final Volume : 50mL
5mL Tris 1M
1mL 0.5M EDTA
44mL MiliQ
100 ml TES:
10 ml TE 10x
25 ml 2% SDS (dissolve in MiliQ)
65 ml H2O (MiliQ)
Preparation:
Final Volume : 100mL
2.5 ml 20% SDS (if you see crystals on it heat it a bit with warm tap water until it dissolve , WITHOUT VORTEXING IT)
10 mL TE 10x
87.5mL MiliQ
Weight 500-600mg of cell pellet
0.793 g from ylic133_2
0.657 g from ylic136_1
0.643 g from ylic136_2
Wash with 30 ml H2O, spin down again and aspirate
Wash two times until the supernatant is clear.
Resuspend in 3 ml Sorbitol Solution +10 \(\mu\)l R-Zymolase.R-Zymolyase contains 5 U/\(\mu\)l Zymolyase.
Incubate at 37\(C\) for 1 hour. Time step can be extended, cell lysis with Zymolyase is delicate and longer incubation does not adversely affect DNA quality
at @ 13:20 the incubation started. I will leave it for ~2hours.
Distribute ~1 ml of sample into 1.5 ml Eppendorf tubes.
at 15:15
Spin down at 3000g and aspirate
Resuspend each pellet in 380 ul TES
Pipette in and out and vortex after
Incubate at 65\(C\) for 30 min
at 15:40 - 16:10
Add 80 \(\mu\)l of 5M KAc to each tube, mix by inversion
Incubate tubes on ice for 15 min
Spin down sample 3 minutes at 12.000g at RT
Transfer supernatant to a clean 1.5 ml Eppendorf tube
Add 1 ml 100% EtOH (4\(C\))
Incubate on ice for 1 minute
Spin down at 12.000g for 3 minutes at RT
Remove supernatant
Add 1 ml of 70% EtOH (4\(C\)) and gently resuspend pellet by pipetting
Incubate on ice for 1 minute
Spin down at 12.000g for 3 minutes at RT
Dry pellet at 65\(C\) for 5 minutes
Resuspend pellet in 150 \(\mu\)l of H2O
20082020
The nanodrop give non trustable results because this prep is very dirty. In my case when measured give like ~1 ug/ul and I have 300ul so I would have like 300ug of DNA , which is a lot! The expected amount for 500mg pellet is from 40-60ug DNA. See the recommendations from Benoit
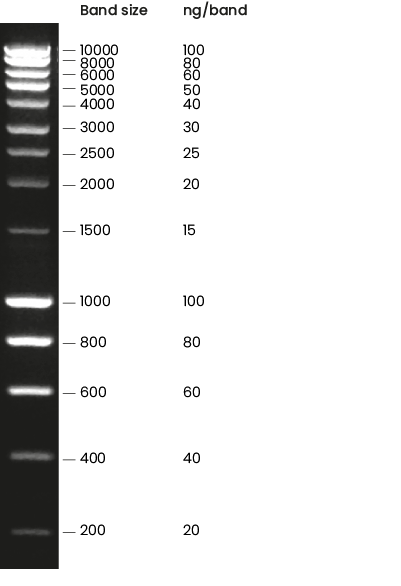
Gel quantification with the ladder intensity at 10:35.
10X dilution sample from the supernatant (without vortexing it)
 {width=40%}
{width=40%}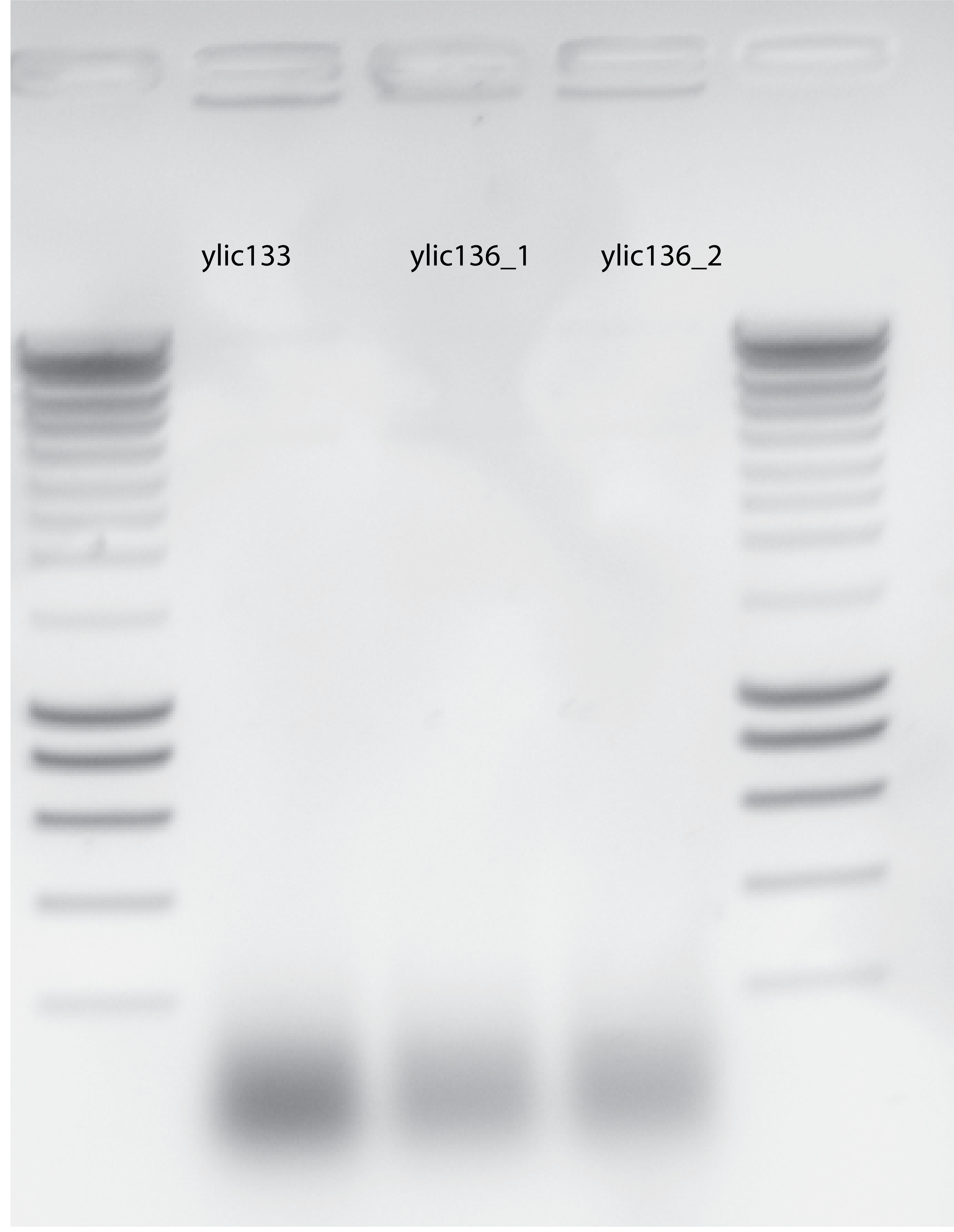 { width=50% }
{ width=50% }very light the upper bands that indicate a very low concentrated genomic DNA, and a very high concentration of RNAs at the bottom.
21082020
Try the protocol for DNA extraction from Benoit, to see the differences with this one.
Protocol from HERE:
A 500 mg cell pellet is resuspended with 500 \(\mu\)l Cell Breaking Buffer (2% Triton X-100, 1% SDS, 100 mM NaCl, 100 mM Tris-HCl pH8.0, 1 mM EDTA) and distributed in 280\(\mu\)l aliquots.
I used ylic133 and ylic136_1 pellet. Both weighted 516mg.
Cell Breaking Buffer recipe with the available components for a 50mL final volume:
1mL 2% Triton
0.5mL 1% SDS
1.25mM of 4M NaCl
5mL 1M Tris
100uL 0.5M EDTA
42.15 mL MiliQ
200 \(\mu\)l Phenol:Chloroform:Isoamylalcool 25:25:1 and 300 \(\mu\)l glass 0.4-0.6mm unwashed glass beads are added to each aliquot.
safety measure: Pipette the Phenol:Chloroform:Isoamylalcool 25:25:1 in the Fume Hood!!!
I took the beads from the Genomic DNA extraction kit. (6 in total, 3 aliquots per strain)
Samples are vortexed for 10 min at 4C using a Disruptor Genie from Scientific Industrial (US Patent 5,707,861).
200 \(\mu\)l TE are added to each lysate, which are then centrifuged for 5 min at 16100x g, 4C.
The upper layer (~400 \(\mu\)l) is transferred to a fresh tube, 2.5vol 100% EtOH are added and the sample mixed by inversion.
DNA is pelleted for 5 min at 16100x g, 20C.
The supernatant is removed and the pellets resuspended in 200 \(\mu\)l RNAse A 250 \(\mu\)g/ml for 15 min at 55°C, 1000 rpm on a Thermomixer comfort (Eppendorf).
I did not pipetted in and or vortex when I added the RNAase A, I just put the eppi with tape in a plastic cap that was shaking at 250rpm at 55C (incubator from the lab). After it finished I vortexed the tubes since the protocol said it should shake at 1000rpm . There was still a pink pellet like flake hard to resuspend again with ethanol.
2.5 vol 100% EtOH and 0.1vol NaOAc 3 M pH5.2 are added and the samples mixed by inversion.
DNA is pelleted by centrifugation for 5 min at 16100x g, 20C.
The pellets are washed with 70% EtOH under the same conditions, the supernatant removed completely, and the pellets let dry for 10 min at 37C.
The pellets are resuspended in a total volume of 100 \(\mu\)l water for 10 min at 55C, 700 rpm on a Thermomixer comfort (Eppendorf).
when I pipette in and out I could be able to resuspend entirely the pellet like flake.
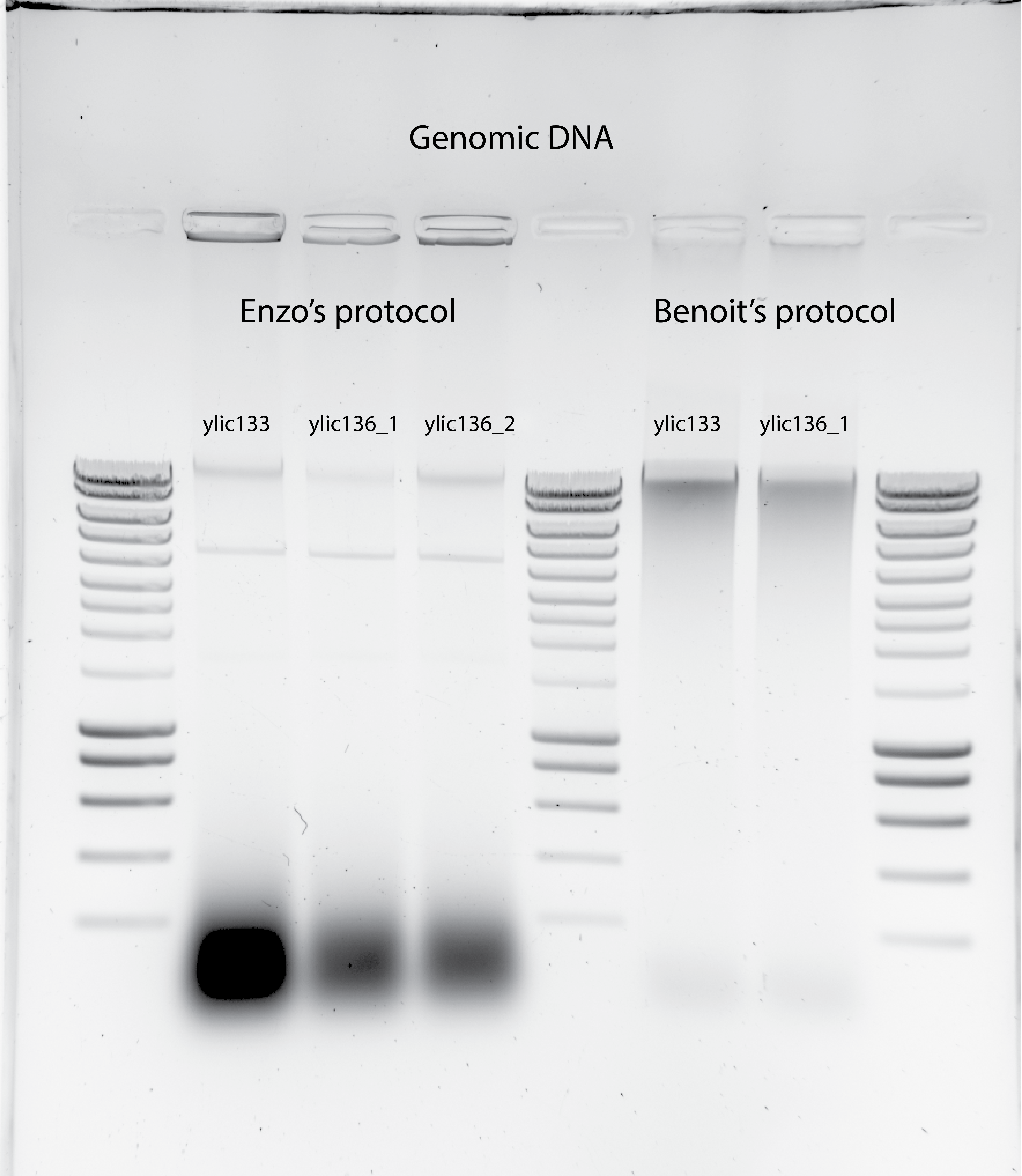
DNA is run on a 0.6% 1X TBE agarose gel against a standard 1 kb GeneRuler, and quantified using FiJi. 500mg cell pellet should yield approximately 20-60 \(\mu\)g DNA. Always quantify on gel. Absorbance reading of this sort of “dirty” prep yields entirely meaningless results!!!
at 15:25 I put the gel on 1% agarose 120V for 30 mins.
I added non diluted samples from Enzo’s protocol without dilution. I took only the supernatant and trashed the pellet.
 {width=30%}
{width=30%}No RNAs at the bottom :)
DNA quantification:
Open the gel in Image J and Measure the integrated intensity of the bands of interest.
Select one band to calibrate , in this case, I used the 100ng band .
Measure the intensity of the background. (empty region in the gel.)
\(\frac{I_{band}-I_{back}}{I_{100ng}-I_{back}}=N_0*I_{100ng}\)
For the band of ylic133 (I took a bit larger section due to the smear) the number was \(4.5*I_{100ng}=450ng\) but because I loaded 10uL then I have roughly 45ng/uL of DNA. For 300uL of DNA I end up with 13.5ug.
For the band of ylic136_1 (I took a bit larger section due to the smear) the number was \(3.5*I_{100ng}=350ng\) but because I loaded 10uL then I have roughly 35ng/uL of DNA. For 300uL of DNA I end up with 10.5ug.
For the bands from Enzo’s protocol I got less than 3ng/ul.
35.4. Results#
At the end of the protocol still there were some solid stuffs forming a kind of pellet . Also still the pink stain stayed in the sample. It seems this is OK for the type of dirty prep it is. Usually it can be centrifuged again and store the supernatant as the DNA. (Enzo advice)
The protocol from Benoit gave better results. Though I got for 300uL of DNA I have 13.5 ug and 10.5 ug of DNA which is a bit slower that what they indicate which is from 20 to 60 ug.
35.5. Next steps#
Do the DNA genomic extraction of ylic136_2 as well following Benoit protocol.
24082020: ylic136_2 Genomic DNA extraction.
what I did differently:
In step 7 I did pipette a bit more in and out , despite the pellet is really hard to resuspend completely. (Maybe that was what caused a bit more RNAs at the end than previous strains , look at the gel)
In step 8 I added 0.1V of NaOAc of 200ul+500ul (the volume with RNAse and EtOH), so 70ul in total , NOT 20ul as previously. Because I did 0.1V of 200ul (Volume with the RNAseA )
DNA concentration, quantified on gel, using ImageJ: 72.5ng/uL , which lead to a final concentration of ~21ng. :)
 {width=30%}
{width=30%}
35.6. Conclusion#
The DNA extraction was OK to continue for the next steps.
- 1
Protocol courtesy of Enzo Kingma