
Title : Digestion and Ligation Test for new SATAY DNA
Contents
31. Title : Digestion and Ligation Test for new SATAY DNA#
31.1. Date#
19102020-21102020
31.2. Objective#
To test if the new DNA extraction from the pellets of WT, \(\Delta\)nrp1_1, \(\Delta\)nrp1_2 is OK to continue with the big batch for sequencing.
31.3. Method#
Try 10X downscaled protocol
DNA concentration of 0.2ug for digestion
WT (~50ng/ul) = 4ul of DNA
\(\Delta\)nrp1_1 (~50ng/ul) = 4ul of DNA
\(\Delta\)nrp1_2 (~40ng/ul) = 5ul of DNA
DNA (uL) |
Buffer NIaIII(uL) |
NIaII(uL) |
MiliQ |
Buffer DPNII(uL) |
DPNII(ul) |
MiliQ |
|
|---|---|---|---|---|---|---|---|
WT |
4 |
0.5 |
0.5 |
0 |
0.5 |
0.5 |
0 |
dnrp1_1 |
4 |
0.5 |
0.5 |
0 |
0.5 |
0.5 |
0 |
dnrp1_2 |
5 |
0.7 |
0.5 |
0.8 |
0.7 |
0.5 |
0.8 |
control buffer DPNII |
1 |
0.2 |
0 |
0.8 |
0.2 |
0 |
0.8 |
control buffer NIaIII |
1 |
0.2 |
0 |
0.8 |
0.2 |
0 |
0.8 |
31.4. Results#
31.4.1. Digestion#
1ul loaded DNA
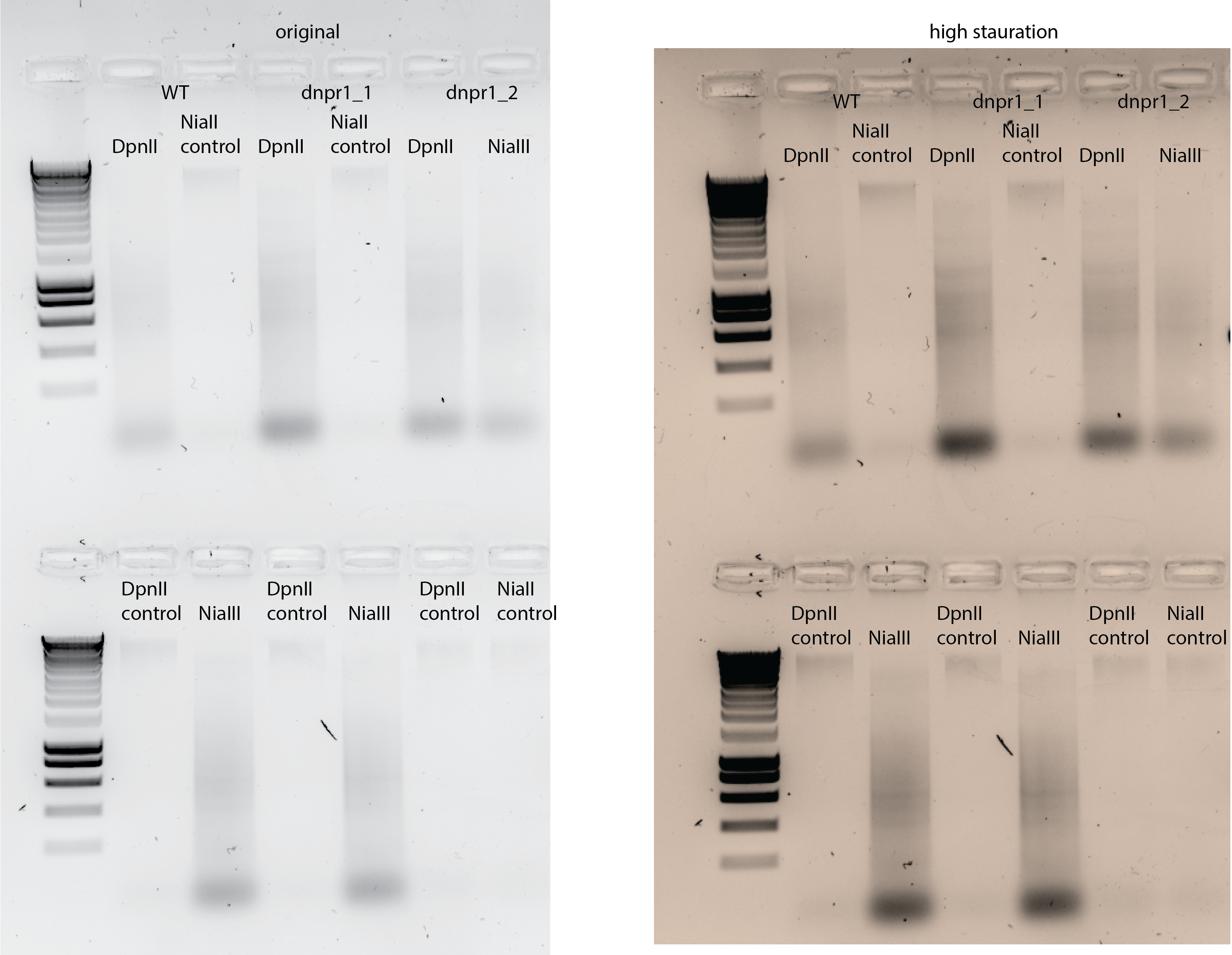
31.4.2. Ligation#

I noticed that there is less smear intensity compared with the intensity of the bands for the unstranposed plasmid.
I suspect that this could be due to the fact that I did not use Non-sticky tubes RNA free…
I asked Benoit in the forum about that , to see if this is OK. See HERE
31.5. Conclusion#
What Agnes replied to me in the SATAY forum ! :
The non transposed vector will always be present but you are right that the corresponding bands are abundant in your PCR products.I would not send these PCR products for sequencing and I see two issues that need addressing first: the regrowth of the library and the down scaling of the starting material for your test run
1-The intensity of the untransposed vector band depends on the reseeding/growing of the library in SC -ADE Glucose. This steps amplifies the transposed ADE+ cells while diluting the cells that contain the non transposed vector. At the end of the regrowth ( =harvest), the proportion of ADE+ cells over the total number of cells reaches ~50% or higher. This can be assessed by plating aliquots of the regrown culture on SC complete, SC -URA and SC -ADE plates.
It seems that the regrowth of the library hasn’t worked well. My feeling is that this is the main culprit. Could you describe how this step was carried?
2-That said, down scaling the gDNA will result in reducing the complexity of the library, which will reflect in your PCRs. Indeed, 2microg of gDNA correspond to ~150millions genomes. 0.2microg is 15millions. If your library contains for example 3millions independent clones (which isn’t great for a liquid library but yours was done plate, right?), then 0.2microg gDNA contain at most 5 copies of each transposition event. That is if 100% of the cells are ADE+ at the time of the harvest, which is never the case. Also, it is pretty much guarantied that the ligation is not 100% efficient. In the end plenty of the transposed template might never be amplified.
So my advice to get an accurate view of the quality of the final PCR product is to digest and ligate 2microg gDNA, as you would for the real experiment. Then I would PCR 1/10 of the ligated product (and include a no template control). If that looks good, you can just PCR the rest, pool the products and purify 100microl.
Here is a general tip for when you are ready to PCR the entire library.
After precipitation and resuspension of the ligation do not transfer the product to a new tube (I typically resuspend in 105microl MQ water). Instead add the PCR mix directly to the template, mix well and only then aliquot the PCR reactions. The template can sometimes flake a bit so this ensures that it all goes into the PCR mix (and the PCR buffer helps getting it in suspension). If you want to try the PCR first, it is totally fine (I have done it). Just make sure your template DNA is well resuspended.